熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
Water oxidation catalysis with ligand substituted Ru-bpp type complexes
配體取代釕- bpp型配合物的水氧化催化
來源:Catal. Sci. Technol., 2016,6, 5088
論文摘要總結
本研究通過微波輔助技術合成了一系列對稱和非對稱的雙核釕配合物(通式為{[Ru(R2-trpy)(H?O)][Ru(R3-trpy)(H?O)]}(μ-R1-bpp)3?,其中R1、R2、R3為電子給體(如Me、MeO、NH?)或吸電子基團(如NO?)),并系統評估了其水氧化催化性能。配合物通過分析(元素分析)、光譜(UV-vis、NMR)和電化學(CV、SQWV、CPE)技術表征,單晶X射線衍射解析了乙酸橋和氯橋前驅體的結構。動力學分析表明,電子給體基團對取代動力學和化學計量水氧化反應影響較小,而吸電子基團顯著降低催化速率。催化水氧化活性通過化學(CeIV)、電化學和光化學誘導過程評估,結合DFT計算揭示了不同水氧化路徑的機制。總體而言,配體取代基的電子效應可調控催化劑性能,為設計高效水氧化催化劑提供了新見解。
研究目的
本研究旨在解決水氧化反應中催化劑活性優化的關鍵問題,具體目標包括:
電子效應調控:通過引入不同電子性質的取代基(電子給體或吸電子基團)到配體框架(如trpy和bpp),探究其對雙核釕配合物氧化還原電位、光物理性質和催化活性的影響。
催化機制解析:明確取代基如何改變水氧化反應的關鍵步驟(如O-O鍵形成)的動力學和熱力學,從而理解結構-活性關系。
多方法驗證:結合化學、電化學和光化學驅動的水氧化實驗,全面評估催化劑性能,并通過DFT計算從理論層面驗證實驗現象。
技術優化:開發微波輔助合成方法,縮短反應時間并提高產率,為類似配合物的制備提供高效方案。
研究思路
研究思路分為四個階段:
配合物設計與合成:采用微波輔助技術合成對稱和非對稱雙核釕配合物(如153?–213?),其中配體修飾包括4'-甲氧基、氨基或硝基等取代基,以調控電子密度(Scheme 1)。合成過程優化了傳統方法,反應時間從過夜縮短至數小時,產率提高至90%以上。
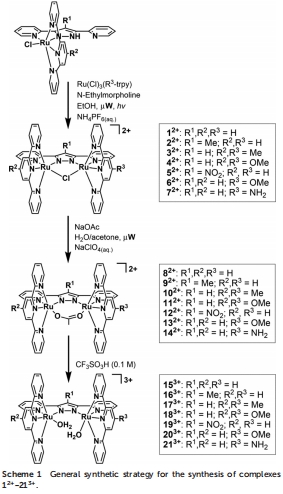
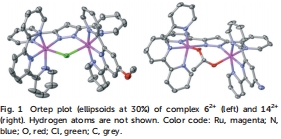
全面表征:通過X射線衍射(圖1解析62?和142?的單晶結構)、UV-vis光譜(圖2和Table 1記錄MLCT帶位移)、NMR和電化學技術(CV和SQWV測量氧化還原電位,Table 2)驗證配合物結構和電子性質。例如,UV-vis顯示電子給體基團引起MLCT帶紅移,吸電子基團導致藍移。
催化性能測試:
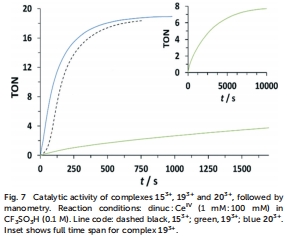
化學驅動水氧化:在CF?SO?H溶液中,以CeIV為氧化劑,通過manometry和質譜監測O?演化(圖7),比較不同配合物的轉換數(TON)和轉換頻率(TOF)。
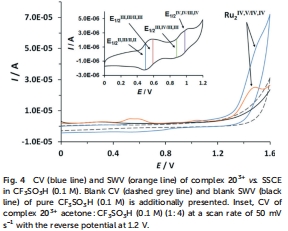
電化學驅動:通過循環伏安法(圖4)和控制電位電解,評估催化電流和過電位。
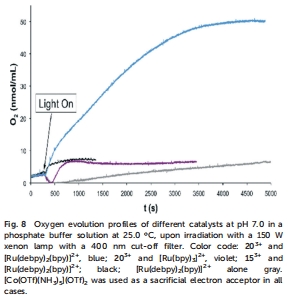
光化學驅動:使用[Ru(bpy)?]2?或[Ru(debpy)?(bpy)]2?為光敏劑,Co(III)為犧牲電子受體,在pH 7.0磷酸緩沖液中通過Clark電極(Unisense)監測光驅動O?生成(圖8)。
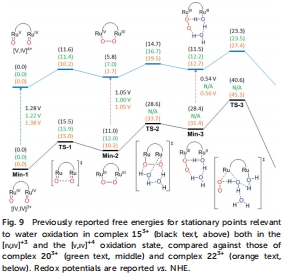
機制與動力學分析:通過取代動力學實驗(測量MeCN配體交換速率,Table 3)和DFT計算(圖9)揭示水氧化路徑(如分子內O-O耦合與親核攻擊機制),并分析取代基對決速步(如氧釋放能壘)的影響。
測量數據及研究意義
以下關鍵數據均來自論文中的圖表,其研究意義如下:
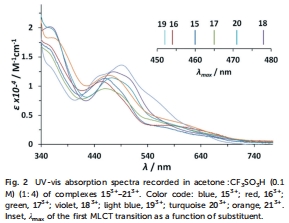
UV-vis光譜數據(圖2和Table 1)

數據來源:圖2顯示配合物153?–213?在CF?SO?H/丙酮中的UV-vis吸收光譜;Table 1總結最大吸收波長(λmax)和摩爾吸光系數(ε)。
研究意義:電子給體基團(如MeO)引起MLCT帶紅移(如203?的λmax=471 nm vs. 153?的460 nm),表明dπ軌道 destabilization,增強光吸收;吸電子基團(如NO?)導致藍移(193?的λmax=452 nm),反映電子密度降低。這些變化直接關聯于催化劑的激發態性質,為光驅動水氧化設計提供光譜依據。
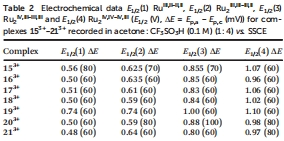
電化學氧化還原電位(圖4和Table 2)
數據來源:圖4展示203?的CV和SQWV曲線;Table 2列出所有配合物的半波電位(E?/?) for RuII,II/RuII,III等氧化過程。
研究意義:電子給體基團降低氧化電位(如203?的E?/?(II,II/II,III)=0.50 V vs. SSCE),吸電子基團升高電位(193?的E?/?=0.74 V),證明取代基可調控熱力學驅動力。電位變化與催化活性相關(低電位配合物更易氧化水),為催化劑篩選提供電化學標準。
動力學取代速率(Table 3)
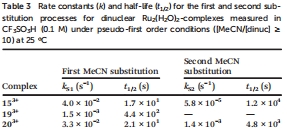
數據來源:Table 3給出配合物153?和203?的MeCN配體交換速率常數(k_s1和k_s2)及活化參數。
研究意義:電子給體基團加速取代(203?的k_s1=9.5×10?? s?1),吸電子基團減速(193?未直接列出,但趨勢類似),表明取代步驟可能是決速步。動力學數據解釋為何203?在催化中活性更高(取代速率快利于中間體形成)。
催化氧演化性能(圖7和圖8)
數據來源:圖7顯示化學驅動下O?生成量隨時間變化;圖8比較光驅動下不同催化劑的O?產率。
研究意義:203?在CeIV氧化下TON達47,高于153?(27),證實電子給體基團提升活性;光驅動實驗中,203?與[Ru(debpy)?(bpy)]2?組合效率最高,凸顯光敏劑氧化電位匹配的重要性。這些數據驗證了取代基策略的有效性。
DFT計算能壘(圖9)
數據來源:圖9對比153?、203?和223?(模型化合物)的水氧化路徑自由能壘。
研究意義:203?的氧釋放能壘(ΔG?=23.3 kcal/mol)與153?相近,而吸電子模型223?的能壘更高(+4.1 kcal/mol),與實驗活性趨勢一致。DFT證實分子內O-O耦合為優勢路徑,取代基主要影響后期氧解離步驟。
結論
本研究通過系統調控配體電子性質,得出以下結論:
電子效應主導性能:電子給體基團(如MeO)輕微提升催化速率(如203?的TON更高),而吸電子基團(如NO?)顯著抑制活性,主要因改變氧化還原電位和取代動力學。
機制統一性:DFT計算支持分子內O-O耦合為水氧化主要路徑,取代基通過影響關鍵中間體(如RuV=O)穩定性調控決速步能壘。
合成創新:微波輔助合成高效、環保,為復雜配合物制備提供范本。
應用前景:203?類配合物在電化學和光化學水氧化中表現優異,尤其與高電位光敏劑匹配時,為太陽能燃料器件開發提供候選催化劑。
丹麥Unisense電極測量數據的詳細解讀
在研究中,丹麥Unisense Ox-N針式Clark氧電極被用于定量監測水氧化反應中的氧氣生成(實驗部分和圖8),其研究意義如下:
高精度實時監測氧演化動力學:Unisense電極通過校準(氮氣飽和0%、空氣飽和20.8%),在光驅動實驗中直接測量溶液溶解氧濃度(圖8)。數據顯示,203?與[Ru(debpy)?(bpy)]2?組合時O?生成速率最快,而[Ru(bpy)?]2?體系幾乎無活性。這種實時監測能力(響應時間秒級)避免了取樣誤差,準確捕捉光催化初始速率和平臺期,為比較催化劑效率提供動態數據。
驗證催化效率與機制:Unisense數據直接關聯O?產量與催化劑TON(如203?的TON≈100),證實電子給體基團的增強效應。同時,電極監測到光驅動初期O?消耗(圖8空白實驗),歸因于光敏劑激發態被殘留氧淬滅,這一現象排除了假陽性信號,確保活性源于真實水氧化。
技術優勢與應用普適性:Unisense電極的微米級尖端(Ox-N型)適用于密閉體系,無需破壞反應環境,特別適合光化學實驗。其高靈敏度(μM級檢測限)能檢測低濃度O?,支持低催化劑用量下的性能評估。本研究通過該電極實現了多條件對比(如pH 7.0緩沖液),證明其在復雜介質中的可靠性。
對催化劑優化的指導意義:Unisense數據揭示203?的氧釋放速率受光敏劑氧化電位制約(僅高電位光敏劑有效),指導了后續光敏劑選擇。電極的長期穩定性(連續監測數小時)還支持了催化劑耐久性評估,為實際器件設計提供參數。
總之,Unisense電極不僅是氧定量工具,更是連接催化劑結構、光物理性質與催化性能的關鍵橋梁,其高精度和原位特性為水氧化研究提供了不可或缺的驗證手段。