熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
結果
甲蟲內部解剖結構
圖1a顯示了O.disjunctus腹腔內的背側腸道排列。FG位于頭部和部分胸腔內(未解剖)。盤繞的MG、AHG和PHG位于腹腔內。盤繞的MG(圖1b)從胃盲囊延伸到馬氏管。向后,腸道突然變寬成一個囊袋區域,即AHG,它位于一層氣管管上方(不可見);細長的PHG緊鄰AHG后方,同樣被氣管管包圍。整個背側腹腔被一層厚厚的角質層覆蓋,并由一對翅膀(已移除)保護,這對翅膀鎖在一起,僅在飛行時打開,而該物種很少飛行。整個腸道的典型長度約為11厘米,FG是最短的區域,MG是最長的區域。
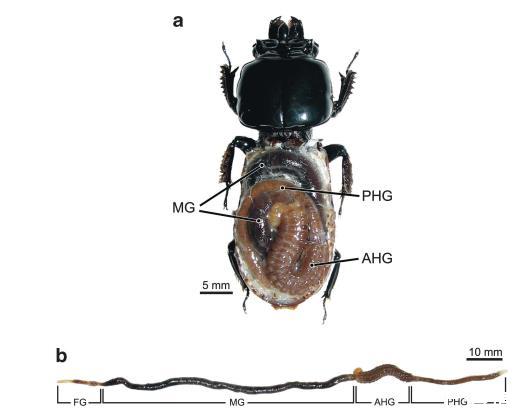
我們用于陣列分析的四只解剖甲蟲的腸道片段重量均勻:FG=13.3±2.1mg,MG=263.5±37.9mg,AHG=125.5±21.4 mg,PHG=47±6.5mg。所有單個腸道片段均產生至少200 ng總(原核和真核)DNA/RNA,用于PCR擴增和cDNA合成。所有腸道區域的細菌16S rRNA基因擴增均成功。然而,古菌16S rRNA基因僅在AHG中成功擴增。在另一組甲蟲中測量了pH,每個腸道區域的平均pH為:FG=7.44±0.21,MG=8.38±0.12,AHG=7.21±0.05,PHG=6.89±0.18。
氧氣分布
黑蜣科甲蟲腸道呈現出一組復雜的微環境,因為存在至少四個形態上不同的區域(圖1b)。為了更好地理解微生物組成或活性的任何區室化,使用微電極測量了每個腸道區域內不同位置的O2分布(圖2)。在每次測量期間,使用顯微操縱器將尖端驅動到腸道壁表面,并使用數字顯微鏡進行監控,并將其保持在該位置,直到測量到穩態的O2讀數(約1分鐘)。在每種情況下,一旦電極尖端穿透腸道壁,氧氣濃度迅速下降,出現陡峭的梯度,直到觀察到氧氣完全耗盡。厭氧區域不僅在AHG中,而且在所有四個腸道區域中都占據了體積優勢(圖2)。
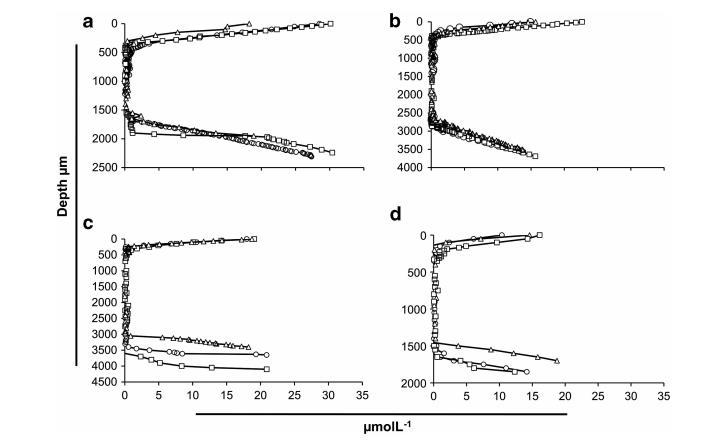
在許多白蟻中,類似的厭氧區僅限于后腸囊。將電極尖端連續移動穿過甲蟲腸道直到到達對側,顯示出的O2分布與進入腸道時獲得的分布呈鏡像。通過測量Nardi等人報道的圖像中AHG和PHG的腸壁厚度,并根據本文確定的O2分布,所有腸道區域的特征都是從微需氧條件到管腔中厭氧條件的快速轉變。考慮到腸壁厚度<100μm,FG和MG中氧氣可利用范圍>300μm,而AHG和PHG分別約為50μm和280μm,其中腸壁厚度分別為200μm和110μm。
腸道區域群落組合
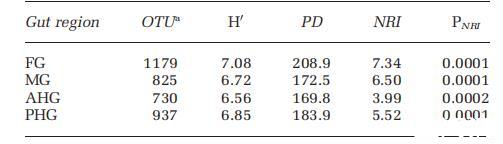
對于群落組成分析,當一個分類群在至少三個重復(每個腸道區域四個重復)中高于檢測閾值(pf>=0.90)時,才被認為存在。基于這些標準,FG的豐富度最高,其次是PHG,然后是MG和AHG。在一系列pf閾值(0.90-1.0;)下觀察到這種模式,其中豐富度模式在R為0.98和0.99之間相關。PD和香農多樣性指數支持了豐富度測量所暗示的模式,即FG是多樣性最高的腸道區域,其次是PHG、MG和AHG。
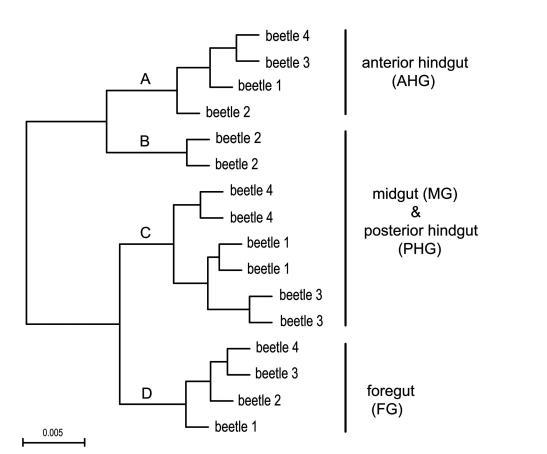
所有16個腸道區域的層次聚類(圖3)表明,FG和AHG形成了兩個不同的簇,而MG和PHG群落沒有被解析,而是按單個甲蟲聚集在一起。特定分類群的相對豐度(探針集強度)在不同腸道區域之間也存在統計學差異。檢測到的分類群的系統發育樹顯示了四個腸道區域中細菌分類群的平均相對豐度(白色到紅色的梯度環表示相對豐度增加;圖4)。
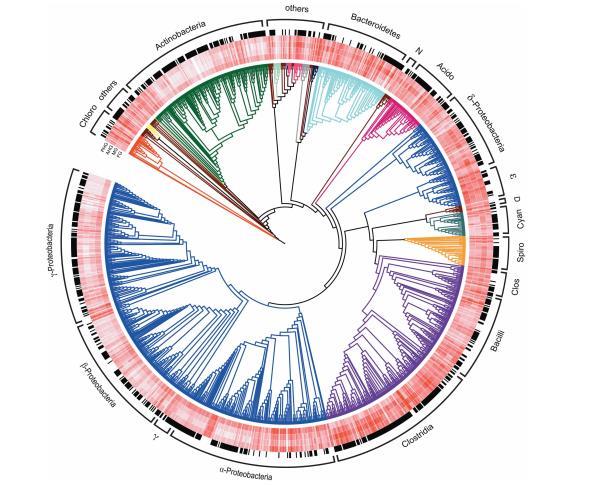
最外層的環(黑色)顯示了在至少兩個腸道區域之間相對豐度具有統計學顯著差異的分類群。檢測到的大多數分類群(75%)在不同腸道區域之間的分布顯示出顯著差異。
為了檢驗每個腸道區域作為不同環境過濾器的假設,我們評估了每個腸道區域的系統發育聚類。系統發育聚類由NRI表示,其中增加的正值代表群落按系統發育親緣關系更聚集,而增加的負值代表更分散的系統發育信號。我們的結果表明,每個腸道區域內的群落都顯著聚集,FG和PHG區域顯示出最大的聚集性(P<0.001;表1)。
O.disjunctus消化道的相鄰區域顯示出其細菌群落組成的變化(圖5)。
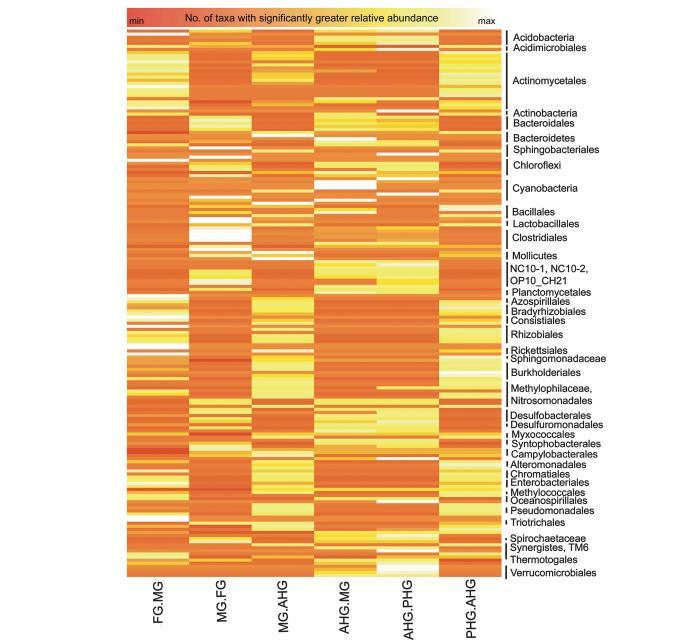
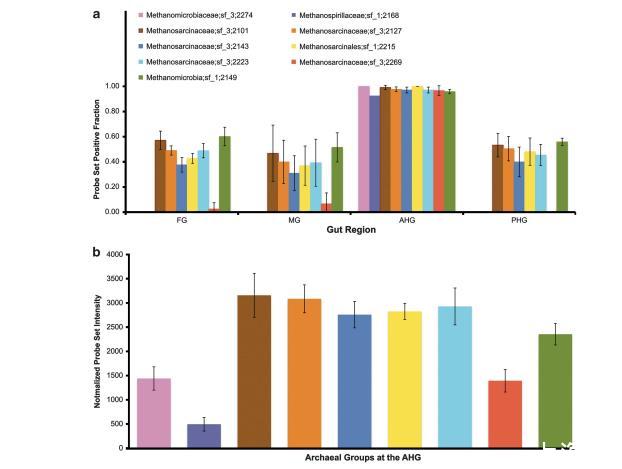
需氧類群如放線菌目(Actinomycetales)、伯克霍爾德菌目(Burkholderiales)、根瘤菌目(Rhizobiales)、著色菌目(Chromatiales)、固氮螺菌目(Azospirillales)、慢生根瘤菌目(Bradyrhizobiales)和亞硝化單胞菌目(Nitrosomonadales)在腸道的兩端(FG和PHG)富集,而在中央區域(MG和AHG)豐度較低。厭氧細菌類群包括擬桿菌目(Bacteroidales)、梭菌目(Clostridiales)、螺旋體目(Spirochaetales)、脫硫桿菌目(Desulfobacterales)、脫硫單胞菌目(Desulfomonadales)、互營桿菌目(Syntrophobacterales)和熱袍菌目(Thermotogales),相對于MG和PHG,在AHG中富集。產甲烷菌,包括甲烷微菌科(Methanomicrobiaceae)、甲烷螺菌科(Methanospirillaceae)和甲烷八疊球菌科(Methanosarcinaceae),僅在AHG中檢測到(圖6a和b)。
通過454焦磷酸測序16S rRNA基因V9區域也評估了與黑蜣科甲蟲不同區域相關的微生物群落。該分析證實了PhyloChip獲得的結果,即微生物類群的分布,即需氧細菌在FG和PHG中更豐富,厭氧細菌在AHG和MG中占主導地位,并且AHG棲息著產甲烷古菌。群落β多樣性的UPGMA(非加權配對算術平均法)聚類顯示出與PhyloChip相似的聚類模式,FG和AHG聚類在分化良好的進化枝中,而MG和PHG聚集在一起,但主要根據甲蟲而不是腸道區域進行聚類,正如PhyloChip分析所觀察到的那樣。
NifH表達和豐富nifH轉錄本的系統發育
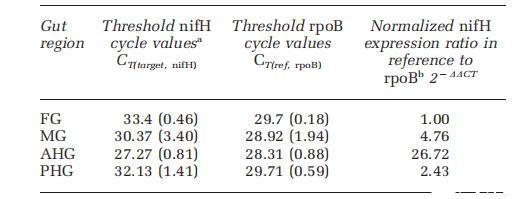
首先通過將甲蟲在15N2頂空環境中孵育12天,然后解剖并對提取的腸道RNA進行整體同位素比質譜分析,來分析固氮作用。所有四個區域都顯示出15N2富集。盡管由于需要較長的孵育時間(約12天)才能達到15N檢測,從這些分析中確認N2固定活性的位置很困難。為了評估每個腸道區域的N2固定潛力,通過qPCR評估了nifH基因表達,并標準化為rpoB基因的表達。在整個腸道中,FG中的表達水平最低;其CT值用作相對定量的校準品(表2)。相對于FG,AHG中的nifH表達高出26.7倍,MG中高出4.7倍,PHG中高出2.4倍。通過克隆和系統發育分析鑒定了N2固定類群。
在所有腸道區域中,優勢類群是一組與Paludibacter propionicigenes的Ni-Fe固氮酶相關(相似性93-96%)的序列。此外,在FG中鑒定出與Clostridium cellobioparum、Azoarcus sp.和Bradyrhizobium japonicum相關的nifH序列,而MG包含與Azorrhizobium doebereineae、Bradyrhizobium denitrificans和Beijerinckia sp.相關的序列。與所有其他腸道區域一樣,AHG中鑒定的大多數序列也與P.propionicigenes相關,部分序列與C.lentocellum相關。PHG顯示出三個主要的nifH序列組,(1)與P.propionicigenes相關的一組,(2)另一個與需氧固氮菌Methylocapsa sp.相關,以及(3)與C.cellobioparum相關的一組。
 相關新聞
相關新聞